
Esta imagem é um trabalho derivado de: 1. LadyofHats derivative work: Gabby8228 ( ) [released into the Public domain], via Wikimedia Commons. *AND* 2. Zephyris:
) [released into the Public domain], via Wikimedia Commons. *AND* 2. Zephyris:  at the English language Wikipedia [GFDL (www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons.
at the English language Wikipedia [GFDL (www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons.
 Este texto está licenciado com uma Licença Creative Commons – Atribuição-CompartilhaIgual 4.0 Internacional.
Este texto está licenciado com uma Licença Creative Commons – Atribuição-CompartilhaIgual 4.0 Internacional.
As proteínas…
Uma proteína é uma molécula resultante da ligação química – ligação peptídica – de vários aminoácidos. À sequência de aminoácidos que compõe esta cadeia polipeptídica (várias ligações peptídicas) chama-se estrutura primária.
![Figura 1. Principais níveis de organização estrutural das proteínas. Esta imagem é um trabalho derivado de: 1. LadyofHats derivative work: Gabby8228 (Main_protein_structure_levels_en.svg) [Public domain], via Wikimedia Commons. *AND* 2. Zephyris at the English language Wikipedia [GFDL (www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons.](http://zerpoii.opentronix.com/wp-content/uploads/2015/07/hemoglobin2.png)
Esta imagem é um trabalho derivado de: 1. LadyofHats derivative work: Gabby8228 (
) [released into the Public domain], via Wikimedia Commons. *AND* 2. Zephyris: at the English language Wikipedia [GFDL (www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons.A hélice-alfa (hélice-α) e a folha-beta (folha-β) constituem as configurações mais frequentes que a proteína pode apresentar. Estas configurações, juntamente com as interacções electrostáticas entre os átomos de oxigénio e os átomos de hidrogénio (ligações por pontes de hidrogénio), constituem as estruturas secundárias. As interacções entre as estruturas primária e secundária designam-se por estruturas terciárias. Por exemplo as interacções entre as estruturas hélice-alfa e folha-beta. A estrutura terciária constitui, em geral, a conformação tridimensional (3D) final de uma proteína – conformação nativa – à qual está associada uma função biológica específica. A estrutura quaternária consiste na interacção de estruturas terciárias que se juntam formando uma nova proteína. Por outras palavras, uma estrutura quaternária é ainda uma conformação final 3D composta por várias conformações finais 3D – uma proteína formada por várias proteínas. A hemoglobina (Figura 1.) é um exemplo de uma estrutura quaternária composta por quatro estruturas terciárias, quatro unidades proteicas (duas hélice-α e duas folha-β) e as correspondentes ligações por pontes de hidrogénio. As várias sub-unidades da hemoglobina interagem originando uma conformação final específica e única, à qual está associada a sua função biológica. (Ler um pouco mais sobre hemoglobina…)
Afinal o que é o folding de proteínas?
Na realidade folding é a palavra inglesa para designar enrolar e dobrar. O folding de proteínas consiste no processo espontâneo de enrolamento e dobragem de uma sequência linear de aminoácidos. Este processo é determinado pela sequência de aminoácidos e das interacções existentes entre eles. Como foi referido acima, a estrutura 3D resultante é única, determina e define a sua função biológica…como o caso da hemoglobina.
Hum…função biológica? Hemoglobina? Isto sugere que estas estruturas específicas podem ser importantes…
Na realidade são muito importantes…sabe-se que podem ocorrer ‘erros’ durante o folding de uma proteína originando uma conformação que não corresponde à chamada conformação nativa. Como se pode antecipar, neste caso, a sua função biológica pode sofrer alterações com consequências significativas, como por exemplo no desenvolvimento e funcionamento de organismos. Voltando ao caso da hemoglobina: uma conhecida alteração de um aminoácido na sequência de aminoácidos, resultante de um ‘erro’ genético, é suficiente para causar uma alteração do processo folding desta proteína, dando origem a uma conformação diferente da conformação nativa. Esta modificação é suficiente para causar alterações da função biológica podendo originar doenças como é o caso da anemia falciforme. Neste tipo de anemia, o folding defeituoso (misfolding) da proteína causa uma alteração da forma das células hemácias, ou glóbulos vermelhos, que passam a ter a forma de foice (daqui o nome anemia falciforme), com eventuais consequências no bem estar do organismo afectado. Existem ainda casos em que uma produção anormalmente elevada de proteínas que ‘foldaram’ incorrectamente se depositam em tecidos ou órgãos. Desta situação pode resultar numa alteração do normal funcionamento (insuficiência) ou na total paragem da actividade dos tecidos ou orgãos. Este é o caso de um conjunto de diversas patologias designadas por amiloidoses (à proteína ‘misfolded’ chama-se amilóide). A proteína em causa e os órgãos e tecidos afectados definem os sintomas e o tipo de amiloidose em questão. Ler mais...
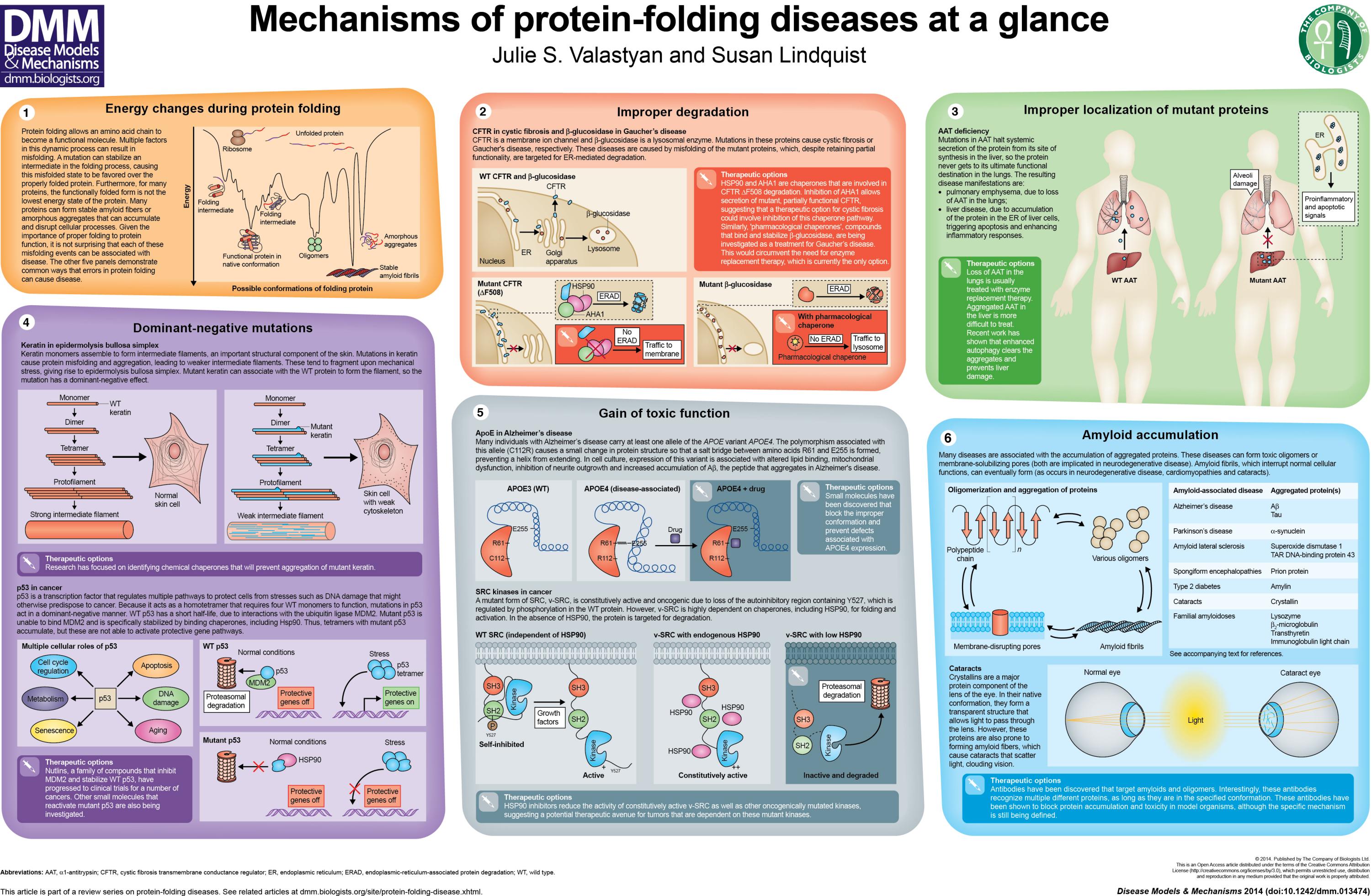
De referir ainda que certos tipos de cancro resultam especificamente de um incorrecto folding das proteínas codificadas pelos designados genes supressores de tumores, ou oncogenes (um pouco mais aqui), em particular responsáveis pela regulação da divisão celular. Sob certas condições, uma pequena alteração genética pode desencadear a produção de proteínas defeituosas que, tendo a sua função biológica alterada (inactivada por exemplo), afecta os mecanismos de controle de divisão ou de morte celular.
O folding de proteínas
Como foi referido acima, uma proteína apenas se torna funcional quando adquire a sua forma 3D (estrutura terciária). Esta estrutura terciária não se consegue determinar de forma simples, de modo que esta informação é conhecida para apenas cerca de 10% das proteínas. Das experiências levadas a cabo por Christian Anfinsen e os seus colegas, nos anos 60, observou-se que: 1. A proteína usada (ribonuclease) pode ser ‘desnaturada’ mudando o ambiente químico, ou apenas aquecendo a solução. 2. O processo de desnaturação pode ser revertido completamente apenas pela reintrodução da proteína no ambiente inicial ou apenas baixando a temperatura. Neste caso a proteína ‘folda‘ para o seu estado 3D natural e biologicamente funcional. Isto levou à construção da Hipótese Termodinâmica, e que valeu a Christian Anfisen o Prémio Nobel da química em 1972 (Nobel Lecture) 1. O processo de folding ocorre de forma espontânea conduzindo a um estado nativo. O estado nativo corresponde ao estado mais estável ou de mais baixa energia, do ponto de vista termodinâmico. 2. A estrutura nativa da proteína é determinada pela sequência de aminoácidos e a totalidade de interacções entre os aminoácidos que a compõem.
Mas…qual é o problema do folding?
Uma questão central no folding de proteínas é saber como é que de uma sequência de aminoácidos como a estrutura primária passa à estrutura terciária, numa escala que pode variar do segundo ao pico-segundos. Cyrus Levinthal propôs que no processo de folding uma proteína colapsa para a sua estrutura nativa sem efectuar uma passagem por todas as configurações possíveis. Esta afirmação baseava-se num simples resultado obtido a partir de uma experiência conceptual. Aqui, será ainda simplificada…(Ler mais…a versão apresentada por C. Levinthal) suponha-se que se tem uma pequena proteína composta por 100 aminoácidos e que cada aminoácido pode encontrar-se em apenas um de dois estados. O número de conformações possíveis acessíveis a uma cadeia de aminoácidos nestas condições é 2100≅ 1030. Considerando que a escala de tempo típica de uma vibração térmica é 1 ps, o tempo que levaria a experimentar o número total de configurações seria 1030 ps ≅ 4.0 × 1010 anos. Ora a idade do universo (estimada) é de cerca de 1.4 × 1010 anos de onde se depreende que a passagem para a estrutura nativa não pode implicar uma passagem por todos os estados possíveis de forma aleatória. Este problema ficou (e é!) conhecido como o paradoxo de Levinthal. Uma abordagem proposta por Levinthal passou por considerar que existem vários estados intermediários que constituem um caminho preferido (único) de folding. Aqui Levinthal propõe que o estado final nativo não corresponde ao estado de energia mais baixo do ponto de vista termodinâmico, ao invés corresponde ao estado de mais baixa energia mas de um ponto de vista cinético. A existência de estados intermediários demonstrou-se não ser necessária com a descoberta (1991) de um proteína que folda sem passar por nenhum estado intermediário. E agora?…Parece que as abordagens de Anfinsen e de Levinthal não se conciliam…O estado nativo corresponde ao estado mais estável ou de mais baixa energia, do ponto de vista termodinâmico ou cinético? A ‘procura’ deste estado é aleatória? Existirá um caminho único? Num próximo artigo falar-se-á de algumas abordagens que desde então têm sido consideradas na resolução do problema de folding. Entretanto, fica aqui a referência a dois projectos que podem ajudar a ‘pragmatizar’ o problema e formas de o tentar resolver: Folding@home e Foldit….e…
…talvez ajudar?
Importa referir que o estudo de um problema tão complexo e exigente como é o folding de proteínas não teria, e não seria, certamente possível sem o paralelo desenvolvimento da capacidade computacional das máquinas que hoje existem. (Será suficiente?…) Como uma das formas que podemos utilizar para ajudar a entender o processo de folding, e como este processo pode ‘falhar’ (misfolding), é recorrendo a métodos computacionais, talvez possamos ajudar o projecto de computação distribuída Folding@home (wiki). ligação para o projecto: (en) https://folding.stanford.edu/ (pt) http://folding.stanford.edu/Portuguese/HomePage

Uma outra abordagem é adoptada pelo projecto Foldit (http://fold.it/portal/). Este projecto apresenta uma panóplia de puzzles que podem ser abordados por qualquer pessoa. O objectivo é ‘ajudar’o computador a aprender as várias estratégias humanas individuais e utilizá-las no estudo do folding de proteínas. Figura 3. Foldit (http://fold.it/portal/).
Este post foi parcialmente motivado por um trabalho realizado no âmbito da cadeira de Física Computacional (com o meu colega Rui P.), leccionado pela Professora Patrícia Faísca, na Faculdade de Ciências de Lisboa (2006). Adicionalmente, o texto foi ainda inspirado em
- O mistério da forma das proteínas (http://cftc.cii.fc.ul.pt/PRISMA/capitulos/capitulo4/modulo4/)
- O mistério da forma das proteínas, Patrícia F.N. Faísca, Gazeta da Física, pp. 34-39, cftc.cii.fc.ul.pt/divulga/Misterio_gazeta.pdf
Outras referências:
- Julie S. Valastyan and Susan Lindquist, Mechanisms of protein-folding diseases at a glance, Dis. Model. Mech. January 2014, doi: 10.1242/dmm.013474 vol. 7 no. 1 9-14.
- http://www.nature.com/horizon/proteinfolding/background.html
- http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1972/index.html